
This might seem blasphemous on a Paleo-focused website, but (gasp) not everyone needs to be strictly gluten-free for their entire lives. Indeed, as discussed in Genetic Adaptations to Neolithic Foods – Understanding Bioindividuality, there is conclusive evidence that humans have adapted genetically, at least somewhat, to the change in diet that occurred with the advent of agriculture, namely the introduction of grains and dairy as substantial proportions of our diet.
 Of course, gluten-containing foods are far from nutritional powerhouses, being nearly devoid of essential nutrients while containing comparable fiber to vegetables and fruit (see Gluten-Free Diets Can Be Healthy for Kids). For many individuals, gluten consumption contributes to impaired gut barrier function (see How Gluten (and other Prolamins) Damage the Gut and What Is A Leaky Gut? (And How Can It Cause So Many Health Issues?)) and gluten may also be linked to weight gain and obesity (see The Link between Gluten and Obesity). There’s also evidence that gluten can alter the microbiome in unfovorable ways, at least in gluten-sensitive individuals, reducing microbial diversity and supporting an inflammatory gut phenotype (see What Is the Gut Microbiome? And Why Should We Care About It?). There’s plenty of evidence that anyone with autoimmune disease or who struggles with their weight will benefit from a nutrient-focused gluten-free diet (see The Autoimmune Protocol). But, it’s a big leap to go from this body of scientific evidence to calling gluten a toxin or stating unequivocally that there’s no place for gluten-containing foods in a healthy diet. Some people can tolerate gluten just fine, provided the overall quality of their diet is high and they’re also following a healthy lifestyle (see The Importance of Nutrient Density and The Paleo Lifestyle). But others (and perhaps the majority of people) react negatively to gluten consumption, which can take the shape of subtle yet insidious symptoms near impossible to link to gluten without doing an elimination diet and challenge (these are the people who have no idea that gluten isn’t doing them any favors until they try a gluten-free diet and discover that their health improves), all the way to the most extreme form of gluten sensitivity, celiac disease.
Of course, gluten-containing foods are far from nutritional powerhouses, being nearly devoid of essential nutrients while containing comparable fiber to vegetables and fruit (see Gluten-Free Diets Can Be Healthy for Kids). For many individuals, gluten consumption contributes to impaired gut barrier function (see How Gluten (and other Prolamins) Damage the Gut and What Is A Leaky Gut? (And How Can It Cause So Many Health Issues?)) and gluten may also be linked to weight gain and obesity (see The Link between Gluten and Obesity). There’s also evidence that gluten can alter the microbiome in unfovorable ways, at least in gluten-sensitive individuals, reducing microbial diversity and supporting an inflammatory gut phenotype (see What Is the Gut Microbiome? And Why Should We Care About It?). There’s plenty of evidence that anyone with autoimmune disease or who struggles with their weight will benefit from a nutrient-focused gluten-free diet (see The Autoimmune Protocol). But, it’s a big leap to go from this body of scientific evidence to calling gluten a toxin or stating unequivocally that there’s no place for gluten-containing foods in a healthy diet. Some people can tolerate gluten just fine, provided the overall quality of their diet is high and they’re also following a healthy lifestyle (see The Importance of Nutrient Density and The Paleo Lifestyle). But others (and perhaps the majority of people) react negatively to gluten consumption, which can take the shape of subtle yet insidious symptoms near impossible to link to gluten without doing an elimination diet and challenge (these are the people who have no idea that gluten isn’t doing them any favors until they try a gluten-free diet and discover that their health improves), all the way to the most extreme form of gluten sensitivity, celiac disease.
So, what makes the difference between the person who feels just fine after a cheat meal of pizza versus the person who is ill for weeks after accidental gluten cross-contamination (and the entire spectrum in between)? There’s growing evidence in the scientific literature pointing to specific gene variants as being the determinative factor in both celiac disease and non-celiac gluten sensitivity.
Let’s start with celiac disease. This is an autoimmune disease impacting an estimated 1 in 133 people, where the body mounts a severe immune attack targeted against the tissues of the small intestine in overresponse to gluten consumption. The damage to the gut barrier is devastating, including something called villous atrophy or pruning, where the villi of the small intestines (microscopic fingerlike protrusive structures designed to increase nutrient absorption, see ) are shortened, leading to severe malabsorption of food nutrients. Villous atrophy is the reason for the classic celiac disease symptoms of severe malnutrition, diarrhea, steatorrhea (found-smelling, fatty, pale stools), and unexplained weight loss with failure-to-thrive or growth stunting in children. However, only about a third of adults with celiac disease actually experience gastrointestinal symptoms. In non-classical celiac disease, the most dominant complaints are other symptoms, such as: chronic fatigue, chronic migraine, peripheral neuropathy (tingling, numbness or pain in hands or feet), unexplained liver damage, osteopenia or osteoporosis, unexplained infertility, depression and anxiety, eczema, and nutrient deficiency syndromes like iron-deficiency anemia. There is also something termed silent celiac disease, in which all the same internal damage is occurring yet there are no outward indicators.
Guide to Nutrivore e-book
Nutrivore is the simple yet revolutionary concept:
Choose foods to meet the body’s nutritional needs!
I’m very excited about the Nutrivore information! Thank you so much for all your research and hard work you put into this kind of information for all of us to use!!
Debbie


There’s also something called non-celiac gluten sensitivity (NCGS). While some medical professionals are still reluctant to accept the existence of this condition, well-designed randomized double-blind placebo-controlled challenge studies have confirmed high prevalence of NCGS in patients with Irritable Bowel Syndrome, who have symptoms disappear completely with avoidance of gluten but re-appear reliably and reproducibly within a few hours or days of gluten challenge. Sufferers of NCGS also have both gastrointestinal and extra-intestinal symptoms, such as brain fog, fatigue, lethargy, skin rash (including eczema), headaches, fibromyalgia-like symptoms (joint and/or muscle pain), carpel tunnel and peripheral neuropathy-like symptoms, depression, anxiety and anemia. The astute observer will note the high degree of overlap between these symptoms and celiac disease, pointing to causal mechanisms in common.

What are those causal mechanisms? Recent scientific research have identified several gene variants that increase your risk of having gluten sensitivity. These have largely been studied in the context of celiac disease, yet it’s now possible to start drawing connections also to NCGS.
What is a Gene Variant?
Before we dive into the specific gene variants linked with celiac disease and NCGS, it helps to explain what exactly a gene variant is!
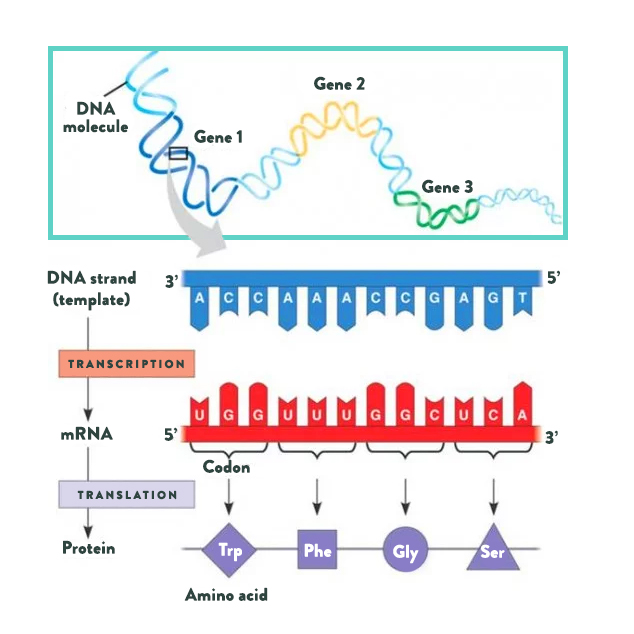 Let’s start with proteins. Proteins, the building blocks of life, are made from long chains of amino acids (the basic structural units of proteins). While approximately five hundred different amino acids have been identified in various life forms, only twenty are used to build every single type of protein in the human body. Three additional amino acids are found in the human body and can be incorporated into proteins after being built (by a chemical reaction called post-translational modification). Various combinations of amino acids are strung together in chains anywhere from twenty to more than two thousand amino acids long. As you can imagine, there are many ways to string twenty different amino acids together. This is how twenty simple building blocks form all the proteins in your body, from the components of the cells of your organs to the hormones that circulate in your blood.
Let’s start with proteins. Proteins, the building blocks of life, are made from long chains of amino acids (the basic structural units of proteins). While approximately five hundred different amino acids have been identified in various life forms, only twenty are used to build every single type of protein in the human body. Three additional amino acids are found in the human body and can be incorporated into proteins after being built (by a chemical reaction called post-translational modification). Various combinations of amino acids are strung together in chains anywhere from twenty to more than two thousand amino acids long. As you can imagine, there are many ways to string twenty different amino acids together. This is how twenty simple building blocks form all the proteins in your body, from the components of the cells of your organs to the hormones that circulate in your blood.
A gene, which is made up of DNA, is basically a detailed map for creating a specific protein within a cell. DNA is made up of a long chain of nucleotides, arranged in base pairs (there are only four nucleotides that make up DNA: adenine [A] which always pairs with thymine [T], and guanine [G] which always pairs with cytosine [C]), which gives it its signature doublehelix structure. Every amino acid is encoded by a unique series of three nucleotides, which is how the DNA map provides the information to create protein. Organelles within the cells “read” the DNA, first “transcribing” DNA into messenger RNA, then “translating” mRNA into protein. Humans have about 23,000 genes, meaning that our cells can produce about 23,000 different proteins.
Nutrivore Weekly Serving Matrix
An easy-to-use and flexible weekly checklist
to help you maximize nutrient-density.
The Weekly Serving Matrix is very helpful! I’ve been eating along these lines but this really helps me know where to focus vs. which foods serve a more secondary role. It’s super helpful and has taken a lot of worry out of my meal planning. Thanks!
Jan

The most common type of genetic variation among people is called a single nucleotide polymorphisms (SNPs, pronounced “snips”). A SNP means that a single nucleotide in a gene is swapped out for a different one. For example, a SNP may replace a cytosine with a thymine (T) in a certain strand of DNA. This then means that a single amino acid is swapped out for a different amino acid in the protein that is encoded by this particular gene. Just changing one amino acid, doesn’t make a huge difference to the protein structure—the same protein is still made—but it can make a subtle difference to the function of that protein, perhaps changing how active an enzyme is or how that protein might bind with its receptor.
When specific SNPs appear in a non-significant proportion of the population, we call it a gene variant. Gene variants are inherited from one or both of your parents. You’re most likely familiar with the C667T variant of the MTHFR gene, which causes reduced activity of the methylenetetrahydrofolate reductase enzyme, which then impacts the methylation cycle (and therefore detoxificaiton pathways, neurotransmitter regulation, etc.). This is an example of one SNP making a huge difference to protein function. Read more in Genes to Know About: MTHFR.
The Nutrivore Collection
Save $10 on Guide to Nutrivore and the Nutrivore Weekly Serving Matrix!
I never realized how important nutrients are and how intricately the body works! I can’t thank you enough for sharing all your knowledge and insights.
Cheryl

HLA-DQ2.5 (rs2187668)
While about 50 different gene variants have been associated with increased celiac disease risk (most of which need more study to fully understand), the strongest association by far is the HLA-DQ2.5 variant of human leukocyte antigen (HLA) gene complex (see also Genes to Know About: Celiac Genes and Genes to Know About: Rheumatoid Arthritis Genes).
The HLA genes encode the major histocompatibility complex (MHC) proteins in humans. The MHC proteins are divided into three classes (creatively named MHC Class I, II, and III). HLA-DQ2.5 is a haplotype (group of genes inherited from a single parent) that is created by the close genetic linkage of two HLA alleles (specific forms of a gene)—DQB1*0201 (the tag SNP is rs2187668, risk allele T) and DQA1*0501—that encode an MHC Class II protein heterodimer (a protein made up of two different proteins).
When an inflammatory cell (specifically, an antigen-presenting cell like a macrophage or a dendritic cell) eats a foreign invader (a process called phagocytosis), a protein fragment from the invader is left on the surface of the inflammatory cell’s own membrane. This protein fragment is bound to a special protein embedded in the cell’s surface called the major histocompatibility complex (MHC) protein. The MHC’s job is to present antigens from foreign invaders the cell has already eaten to the adaptive immune system. When this inflammatory cell meets a type of white blood cell called a helper T cell (either by traveling to the nearest lymph node or by recruitment of lymphocytes to the area by cytokines or complement proteins), the helper T cell recognizes the antigen presented in the MHC and is activated. Once activated, the helper T cells start to divide and produce proteins that activate B cells (the type of cell that produces antibodies) and other types of T cells as well as other immune cells, triggering a response of the adaptive immune system (also called acquired immunity or specific immunity).
The reason why HLA variants are so strongly linked with autoimmune disease is because they affect the function of the MHC proteins. In the case of HLA-DQ2.5, partially digested gluten (or more specifically gliadin peptides that have been deamidated by tissue transglutaminase [tTG]) form a complex with the MHC proteins encoded by the HLA-DQ2.5 genes (as well as tTG), so antigen-presenting cells present gluten (and tTG) to T cells in the same way that they would present a viral or bacterial antigen. The result is activation of gluten-sensitized T-cells and subsequent production of antibodies against gluten (and tTG, see Why Grains Are Bad–Part 1, Lectins and the Gut).
Approximately 95% of celiac disease sufferers have the HLA-DQ2.5 gene variant (almost all of the rest have a related variant called HLA-DQ8). In fact, having HLA-DQ2.5 is the second highest risk factor for celiac disease, the highest is having a close family member with the disease. In fact, people with two copies of HLA-DQ2.5 (one from each parent, also called being a homozygote), have a 20 to 40% lifelong risk for developing celiac disease. HLA-DQ2.5 is also associated with increased risk of dermatitis herpetiformis, type 1 diabetes, Lambert-Eaton myasthenic syndrome (LEMS), Sjögren’s syndrome, autoimmune hepatitis, Moreen’s ulceration, multiple sclerosis, Grave’s disease, Hashimoto’s thyroiditis and systemic lupus erythematosus.
HLA-DQ2.5 is also over-represented in non-celiac gluten sensitivity. While about half of those with diagnosed NCGS have HLA-DQ2.5, only 25 to 35% of the general population has this gene. And the link between HLA-DQ2.5 and NCGS likely is mediated through zonulin production.
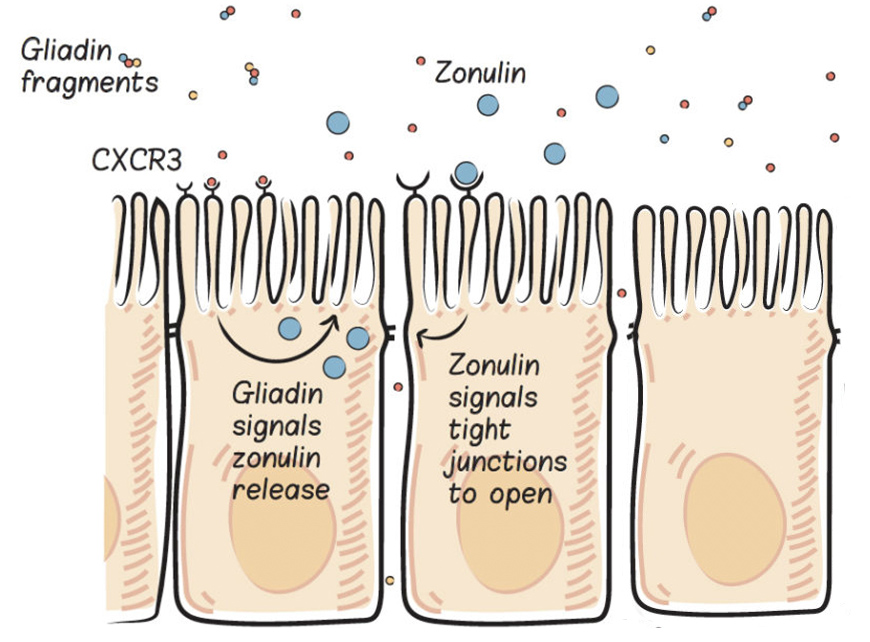
Gluten increases intestinal permeability by stimulating the release of a protein called zonulin. Zonulin (also identified as haptoglobin 2 precursor) used to be thought of simply as a precursor protein, without a distinct function. However, now it is recognized as the only (so far, anyway) extracellular protein that impacts tight junction assembly (dynamic cellular structures that bind adjacent epithelial cells, like those that form the gut barrier, together).
Zonulin is released by gut epithelial cells (called enterocytes) into the gut lumen (inside the gut), where it travels downstream and then binds to at least two different types of receptors in the enterocyte apical membrane (the top part of the enterocyte, which faces into the gut lumen), PAR2 and EGFr. Binding to either of these receptors creates intracellular signaling cascades that displace important tight junction proteins (decreasing the protein-protein interactions that form the tight junction) thereby causing tight junctions to open and intestinal permeability to increase (see What Is A Leaky Gut? (And How Can It Cause So Many Health Issues?) and How Gluten (and other Prolamins) Damage the Gut)
Zonulin may play a normal role in digestion, by opening up the tight junctions to facilitate the absorption of some nutrients into the body. For example, even though glucose is actively transported into enterocytes (via the insulin-dependent GLUT4 transporter), the major portion of glucose is absorbed paracellularly, via increased tight junction permeability. Zonulin may also have a normal role in protecting us from gut pathogens. Zonulin secretion is also stimulated by the presence of enteroinfectious bacteria, which has two effects. First, when the tight junctions open due to zonulin secretion, the hydrostatic pressure gradient flips so that water is secreted into the intestinal lumen (as opposed to being absorbed into the body), which helps to flush bacteria out of the body (in fact, this may explain the high frequency of loose stools and/or diarrhea as a symptom of enteropathogens). Second the increased intestinal permeability exposes the invading pathogen to the immune system, allowing for a more efficient immune response. Interestingly, this might be why gut pathogens are common triggering events for autoimmune disease. And, here’s another way that an unhealthy or unbalanced gut microbiome may be linked to chronic disease: even nonpathogenic strains of E. coli (a Gram-negative normal resident of the human gut, at least in small quantities), causes an increase in intestinal permeability via increased zonulin release. This may represent another important link between leaky gut and the microbiome.
The problem is when there’s too much zonulin.
Too much zonulin causes increased intestinal permeability–yes, leaky gut. And, as you know, leaky gut is linked to every chronic illness in which a potential link has been investigated, hypothesized to be a necessary precursor to autoimmune disease and other conditions (see Which comes first: the leaky gut or the dysfunctional immune system?, What Is A Leaky Gut? (And How Can It Cause So Many Health Issues?), and How Gluten (and other Prolamins) Damage the Gut).
When we consume gluten, it is partially digested (because of its incompatibility with our digestive enzymes) and predictable gliadin fragments are produced. These gliadin fragments bind with a specific receptor in the apical cell membrane of enterocytes, called the chemokine receptor 3 (CXCR3) , which stimulates the release of zonulin into the gut lumen (inside the gut). This happens in everyone, but people with celiac disease are known to have an exaggerated zonulin response to gluten consumption, and this is thought to be a key driver of the disease.
Interestingly, even people without celiac disease but who have HLA-DQ2.5 have an exaggerated zonulin response when they consume gluten. This may be the cause of not only of the gastrointestinal symptoms related to NCGS, but also the systemic systems since increased intestinal permeability has a variety of negative health consequences (again, see What Is A Leaky Gut? (And How Can It Cause So Many Health Issues?)).
CCR3 (rs6441961)

Speaking of chemokine receptor 3, guess what gene encodes that protein? You guess it! CCR3! It turns out that a specific variant in the CCR3 gene (the tag SNP is rs6441961, risk allele T) has also been linked to increased risk of celiac disease (increasing celiac disease risk by about 21%). But, chemokine receptor 3 does a lot more than simply bind with gliadin fragments in the gut to signal zonulin release!
Chemokines are a group of molecules that help to direct leukocyte trafficking, the migration of white blood cells from the circulation into tissue, which occurs during inflammatory processes. And, like all signaling molecules, chemokines achieve their effects through receptor binding. There are many chemokine receptors, with chemokine receptor 3 (CXCR3) known to play a critical role in allergic and other immune-mediated inflammation. CXCR3 can be found in the cell membrane of a variety of different immune cell types, including eosinophils, basophils, dendritic cells, mast cells and Th1 cells (type 1 helper T cells), Th2 cells (type 2 helper T cells), Th17 cells (type 17 helper T cells) and cytotoxic T cells. If you’ve read The Paleo Approach, you recognize Th1, Th2, and Th17 cells as culprits in autoimmune processes. Indeed, increased expression of CXCR3 is not only
linked to celiac disease but also to other autoimmune diseases, including ulcerative colitis, Crohn’s disease, multiple sclerosis, rheumatoid arthritis and systemic lupus erythematosus. While it remains unknown how exactly this particular CCR3 gene variant impacts CXCR3 expression or function, this data implicates an effect on expression, activity or binding affinity, not only leading to increased activation of immune cells but also to exaggerated zonulin release in response to gluten consumption.
 IL21 (rs13119723 and rs6822844)
IL21 (rs13119723 and rs6822844)
Chemokines are a type of cytokine. Cytokines are chemical messengers of inflammation, some are proinflammatory signals and others are anti-inflammatory signals. Cytokines can signal to immature immune cells to develop into specific immune cell subtypes, signal immune cells to divide, and can even impact how responsive various immune cells are to other stimuli. Cytokines include various classes of small proteins, including the aforementioned chemokines as well as interferons, interleukins, lymphokines, and tumor necrosis factors. The IL21 gene encodes a particular cytokine called interleukin-21 (IL-21).
Interleukin-21 is a pleiotropic cytokine, meaning that it has a wide range of seemingly unrelated effects. It is mainly produced by B cells, activated T cells, and natural killer cells and impacts the proliferation (cell division), survival, differentiation (maturation) and function of a broad range of immune cells as well as epithelial cells. For example, IL-21 plays a key role in the differentiation of activated B cells to produce plasma B cells, basically antibody factories, and in the development of Th17 cells, while also blocking the regulatory effects of Treg cells on helper T cell subsets. These actions are why IL-21 is known to promote a variety of autoimmune diseases, including celiac disease, systemic lupus erythematosus, type 1 diabetes, multiple sclerosis, inflammatory bowel disease, and psoriasis. Clinical trials using IL-21 inhibitors are in progress for autoimmune diseases. On the flip side, IL-21 increases survival and activity of cytotoxic T cells, which help to patrol the body for tumor cells and infected cells, which is likely why IL-21 has shown promise as a chemotherapy agent for solid tumors. If you find that dichotomy fascinating, then you’ll definitely want to read The Link Between Cancer and Autoimmune Disease.
Two variants of the IL21 gene (the tag SNP is rs13119723, risk allele G, and the tag SNP rs6822844, risk allele T) are associated with increased risk of celiac disease. Here’s the functional connection: gluten-reactive T cells (as produced in people with the HLA-DQ2.5 gene variant when partially digested gluten binds with CXCR3 receptors) produce IL-21 (and another cytokine called interferon-γ [IFN-γ]) indicating a critical role for IL-21 in the pathogenesis of celiac disease as well as a link, albeit tentative, to gluten sensitivity.

 MYO9B (rs2305764)
MYO9B (rs2305764)
A hallmark of autoimmune disease, including celiac disease, is increased intestinal permeability (see What Is A Leaky Gut? (And How Can It Cause So Many Health Issues?) and How Gluten (and other Prolamins) Damage the Gut), but zonulin is not the only mechanism behind this phenomenon. Another gene variant linked with celiac disease as well as other autoimmune diseases—including systemic lupus erythematosus, rheumatoid arthritis, and type 1 diabetes—is a variant of the myosin IXB (MYO9B) gene (the tag SNP is rs2305764, risk allele G), which encodes a type of motor protein, called myosin-IXB.
Myosins are a huge family of proteins that act as intracellular motors, converting cellular energy (ATP) into movement via interaction with and even remodeling of the cytoskeleton, a collection of actin filaments (also called microfilaments) that provide structure inside the cell and also act like railroad tracks for myosin train cars. Myosins are responsible for a variety of functions including muscle cell contraction, cellular locomotion, phagocytosis (the “eating” of pathogens by certain types of immune cells), and vesicle transport (a method of transportation for proteins within the cell; for example, after a protein is formed in the endoplasmic reticulum, it is encapsulated within a vesicle in the Golgi apparatus, which is then released into the cytoplasm to travel to the organelle or cellular membrane that utilizes that protein). Myosin-IXB is what is called an unconventional myosin, and its function is essential for the gut barrier: it remodels actin within the gut enterocytes and regulates tight junction assembly.
Emerging scientific evidence demonstrates that this particular variant of MYO9B leads to a functional defect in the gut barrier; inflammatory bowel disease patients with this particular gene variant have higher intestinal permeability than those with the wildtype gene. Researchers postulate that this gene variant causes primary impairment of the gut barrier, permitting immunogenic gluten peptides to enter the body and interact with the immune system, setting the stage for gluten intolerance and celiac disease.
Genetic Testing for Gluten Sensitivity
While the gold standard for determining whether gluten is a food that does or doesn’t work for you as an individual remains a gluten elimination (for at least 4 weeks) and challenge, genetic testing may provide some insight, affirmation and confirmation. I have personally done genetic testing for all of the above gene variants, and, no surprise, am heterozygous for the risk allele in 4 out of the 5.
I want to emphasize that while the body of evidence linking these 5 gene variants to celiac disease, and by extension, gluten sensitivity, is fairly robust and includes mechanistic explanations, these are most certain NOT the only genes that contribute to gluten sensitivity. As I mentioned earlier, at least 50 gene variants have been linked with increased celiac disease risk in GWAS (genome-wide association) studies. While the presence of one or more of these gene variants in your DNA certainly indicates that you would benefit from a gluten-free diet, the absence of any of these risk genes does not automatically mean that gluten is your friend.
So, how do you go about determining if you have any of the risk alleles for these genes? Using the RS# in the table below, you can look up which variant you have if you’ve ever done genetic testing through 23andMe, Ancestry or a similar service.
| Gene | RS# | Risk Allele |
| HLA-DG2.5 | rs2187668 | T |
| CCR3 | rs6441961 | T |
| IL21 | rs13119723 | G |
| IL21 | rs6822844 | T |
| MYO9B | rs2305764 | G |
If you have yet to delve into the realm of genetic testing, I recommend checking out the genetic screening panels from MaxGen Labs. I had the honor of meeting the good folks behind MaxGen Labs at last year’s Paleo f(x) conference and I am crazy impressed with their rigor and dedication to serving our community. Where MaxGen Labs truly stands out is that they only test specific genes for which there is substantial evidence of a cause-and-effect relationship with health. Then, they provide you with a detailed report telling you what it means for your day-to-day life. You can also schedule a telephone consult (for an additional fee) to go over your test results with an expert in detail. Max Gen Labs test for the above gene variants linked to gluten sensitivity as part of their MaxFood Panel and, of course, The Works! Panel. You might also wish to check out their MaxFunction Panel (or again, splurge for The Works!) which tests a huge variety of methylation SNPs (see Genes to Know About: MTHFR) as well as your innate detoxification abilities, antioxidants, and immune stimulation.
Citations
Amundsen SS, Rundberg J, Adamovic S, Gudjónsdóttir AH, Ascher H, Ek J, Nilsson S, Lie BA, Naluai AT, Sollid LM. Four novel coeliac disease regions replicated in an association study of a Swedish-Norwegian family cohort. Genes Immun. 2010 Jan;11(1):79-86. doi: 10.1038/gene.2009.67. Epub 2009 Aug 20.
Barbaro MR, et al. The role of zonulin in non-celiac gluten sensitivity and irritable bowel syndrome. Abstract presented at the 23rd United European Gastroenterology Week (UEG Week 2015), October 24–27 2015, Barcelona, Spain.
Bodd M, Ráki M, Tollefsen S, Fallang LE, Bergseng E, Lundin KE, Sollid LM. HLA-DQ2-restricted gluten-reactive T cells produce IL-21 but not IL-17 or IL-22. Mucosal Immunol. 2010 Nov;3(6):594-601. doi: 10.1038/mi.2010.36. Epub 2010 Jun 23.
Carroccio A, Mansueto P, Iacono G, Soresi M, D’Alcamo A, Cavataio F, Brusca I, Florena AM, Ambrosiano G, Seidita A, Pirrone G, Rini GB. Non-celiac wheat sensitivity diagnosed by double-blind placebo-controlled challenge: exploring a new clinical entity. Am J Gastroenterol. 2012 Dec;107(12):1898-906; quiz 1907. doi: 10.1038/ajg.2012.236. Epub 2012 Jul 24.
Cecilio LA, Bonatto MW. The prevalence of HLA DQ2 and DQ8 in patients with celiac disease, in family and in general population. Arq Bras Cir Dig. 2015 Jul-Sep;28(3):183-5. doi: 10.1590/S0102-67202015000300009.
Dema B, Martínez A, Fernández-Arquero M, Maluenda C, Polanco I, de la Concha EG, Urcelay E, Núñez C. Association of IL18RAP and CCR3 with coeliac disease in the Spanish population. J Med Genet. 2009 Sep;46(9):617-9. doi: 10.1136/jmg.2009.067041. Epub 2009 Jun 18.
Hrdlickova B, Westra HJ, Franke L, Wijmenga C. Celiac disease: moving from genetic associations to causal variants. Clin Genet. 2011 Sep;80(3):203-313. doi: 10.1111/j.1399-0004.2011.01707.x. Epub 2011 Jun 13.
Kaukinen K, Partanen J, Mäki M, Collin P. HLA-DQ typing in the diagnosis of celiac disease. Am J Gastroenterol. 2002 Mar;97(3):695-9.
Koskinen LL, Korponay-Szabo IR, Viiri K, Juuti-Uusitalo K, Kaukinen K, Lindfors K, Mustalahti K, Kurppa K, Adány R, Pocsai Z, Széles G, Einarsdottir E, Wijmenga C, Mäki M, Partanen J, Kere J, Saavalainen P. Myosin IXB gene region and gluten intolerance: linkage to coeliac disease and a putative dermatitis herpetiformis association. J Med Genet. 2008 Apr;45(4):222-7. Epub 2007 Dec 12.
Lacotte S, Brun S, Muller S, Dumortier H. CXCR3, inflammation, and autoimmune diseases. Ann N Y Acad Sci. 2009 Sep;1173:310-7. doi: 10.1111/j.1749-6632.2009.04813.x.
Lammers KM, Lu R, Brownley J, Lu B, Gerard C, Thomas K, Rallabhandi P, Shea-Donohue T, Tamiz A, Alkan S, Netzel-Arnett S, Antalis T, Vogel SN, Fasano A. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology. 2008 Jul;135(1):194-204.e3. doi: 10.1053/j.gastro.2008.03.023. Epub 2008 Mar 21.
Maiti AK, Kim-Howard X, Viswanathan P, Guillén L, Rojas-Villarraga A, Deshmukh H, Direskeneli H, Saruhan-Direskeneli G, Cañas C, Tobön GJ, Sawalha AH, Cherñavsky AC, Anaya JM, Nath SK. Confirmation of an association between rs6822844 at the Il2-Il21 region and multiple autoimmune diseases: evidence of a general susceptibility locus. Arthritis Rheum. 2010 Feb;62(2):323-9. doi: 10.1002/art.27222.
Manousou P, Kolios G, Valatas V, Drygiannakis I, Bourikas L, Pyrovolaki K, Koutroubakis I, Papadaki HA, Kouroumalis E. Increased expression of chemokine receptor CCR3 and its ligands in ulcerative colitis: the role of colonic epithelial cells in in vitro studies. Clin Exp Immunol. 2010 Nov;162(2):337-47.
Megiorni F, Pizzuti A. HLA-DQA1 and HLA-DQB1 in Celiac disease predisposition: practical implications of the HLA molecular typing. J Biomed Sci. 2012 Oct 11;19:88. doi: 10.1186/1423-0127-19-88.
Mohan M, Chow CT, Ryan CN, Chan LS, Dufour J, Aye PP, Blanchard J, Moehs CP, Sestak K. Dietary Gluten-Induced Gut Dysbiosis Is Accompanied by Selective Upregulation of microRNAs with Intestinal Tight Junction and Bacteria-Binding Motifs in Rhesus Macaque Model of Celiac Disease. Nutrients. 2016 Oct 28;8(11). pii: E684.
Monsuur AJ, de Bakker PI, Alizadeh BZ, et al. Myosin IXB variant increases the risk of celiac disease and points toward a primary intestinal barrier defect. Nat Genet. 2005 Dec;37(12):1341-4. Epub 2005 Nov 13.
Peluso I, Fantini MC, Fina D, Caruso R, Boirivant M, MacDonald TT, Pallone F, Monteleone G. IL-21 counteracts the regulatory T cell-mediated suppression of human CD4+ T lymphocytes. J Immunol. 2007 Jan 15;178(2):732-9.
Plaza-Izurieta L, Castellanos-Rubio A, Irastorza I, Fernández-Jimenez N, Gutierrez G; CEGEC, Bilbao JR. Revisiting genome wide association studies (GWAS) in coeliac disease: replication study in Spanish population and expression analysis of candidate genes. J Med Genet. 2011 Jul;48(7):493-6. doi: 10.1136/jmg.2011.089714. Epub 2011 Apr 13.
Prager M, Durmus T, Büttner J, Molnar T, de Jong DJ, Drenth JP, Baumgart DC, Sturm A, Farkas K, Witt H, Büning C. Myosin IXb variants and their pivotal role in maintaining the intestinal barrier: a study in Crohn’s disease. Scand J Gastroenterol. 2014 Oct;49(10):1191-200. doi: 10.3109/00365521.2014.928903.
Sánchez E, Alizadeh BZ, Valdigem G, Ortego-Centeno N, Jiménez-Alonso J, de Ramón E, García A, López-Nevot MA, Wijmenga C, Martín J, Koeleman BP. MYO9B gene polymorphisms are associated with autoimmune diseases in Spanish population. Hum Immunol. 2007 Jul;68(7):610-5. Epub 2007 Apr 10.
Sanz Y. Effects of a gluten-free diet on gut microbiota and immune function in healthy adult humans. Gut Microbes. 2010 May-Jun;1(3):135-7. doi: 10.4161/gmic.1.3.11868. Epub 2010 Mar 16.
Spolski R, Leonard WJ. Interleukin-21: a double-edged sword with therapeutic potential. Nat Rev Drug Discov. 2014 May;13(5):379-95. doi: 10.1038/nrd4296. Epub 2014 Apr 22.
Trynka G, Wijmenga C, van Heel DA. A genetic perspective on coeliac disease. Trends Mol Med. 2010 Nov;16(11):537-50. doi: 10.1016/j.molmed.2010.09.003. Epub 2010 Oct 12.
van Heel DA, Franke L, Hunt KA, et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat Genet. 2007 Jul;39(7):827-9. Epub 2007 Jun 10.
van Leeuwen MA, Lindenbergh-Kortleve DJ, Raatgeep HC, de Ruiter LF, de Krijger RR, Groeneweg M, Escher JC, Samsom JN. Increased production of interleukin-21, but not interleukin-17A, in the small intestine characterizes pediatric celiac disease. Mucosal Immunol. 2013 Nov;6(6):1202-13. doi: 10.1038/mi.2013.19. Epub 2013 Apr 10.
Vazquez-Roque MI, Camilleri M, Smyrk T, Murray JA, O’Neill J, Carlson P, Lamsam J, Eckert D, Janzow D, Burton D, Ryks M, Rhoten D, Zinsmeister AR. Association of HLA-DQ gene with bowel transit, barrier function, and inflammation in irritable bowel syndrome with diarrhea. Am J Physiol Gastrointest Liver Physiol. 2012 Dec 1;303(11):G1262-9. doi: 10.1152/ajpgi.00294.2012. Epub 2012 Oct 4.
Wolters VM, Alizadeh BZ, Weijerman ME, Zhernakova A, van Hoogstraten IM, Mearin ML, Wapenaar MC, Wijmenga C, Schreurs MW. Intestinal barrier gene variants may not explain the increased levels of antigliadin antibodies, suggesting other mechanisms than altered permeability. Hum Immunol. 2010 Apr;71(4):392-6. doi: 10.1016/j.humimm.2010.01.016.
Yang G, Zhang B, Huang W, Zhang N, Dong F, Jing L, Wang M, Liu Y, Guo C, Pan H, Wei X, Jing C. Systematic review and meta-analysis of the association between IL18RAP rs917997 and CCR3 rs6441961 polymorphisms with celiac disease risks. Expert Rev Gastroenterol Hepatol. 2015;9(10):1327-38. doi: 10.1586/17474124.2015.1075880. Epub 2015 Aug 8.
Yuan MJ, Wang T. Advances of the interleukin-21 signaling pathway in immunity and angiogenesis. Biomed Rep. 2016 Jul;5(1):3-6. Epub 2016 Apr 27.


 TPV Podcast, Episode 328: The Amazing Health Benefits of Drinking Tea
TPV Podcast, Episode 328: The Amazing Health Benefits of Drinking Tea